

PAST DISCOVERIES BY THE AVML GROUP
Funded primarily by the NIH/NHLBI and AHA, the AVML group has made some important discoveries that:
- delineated the mechanisms by which fluid mechanical forces regulate thrombosis/thrombolysis and inflammation on the vessel wall;
- provided the first evidence that shear stress initiates reactive oxygen species (ROS)-mediated signaling in ECs and investigated the subcellular sources for shear-induced EC ROS production;
- delineated the intracellular mechanisms by which ischemia leads to EC injury, and the intracellular signaling by which ROS induce leukocyte adhesion (inflammation) to post-hypoxic ECs;
- provided the first evidence that mitochondrial ROS (mtROS) regulate shear-induced gene expression;
- delineated the molecular mechanisms by which I/RP leads to mitochondrial and EC dysfunction;
- shed light on the role of bioenergetics in mitochondrial morphology and motility, which determine the EC fate;
- demonstrated the critical role of mitochondrial Ca2+ (mCa2+) uptake, via the Mitochondrial Calcium Uniporter (MCU) channel, in regulating the shear-induced changes in intracellular Ca2+ concentration ([Ca2+]i); and
- assessed the effects of increased MCU activity in mCa2+ signaling in sheared ECs, as well as the effects of prolonged EC exposure to shear stress on MCU complex subunit expression and mCa2+ uptake.
CURRENT RESEARCH PROJECTS BY THE AVML GROUP
Project 1:
Mitochondria-containing endothelial-derived extracellular vesicles as biomarkers and potential therapeutics for cardiovascular diseases (CVD)
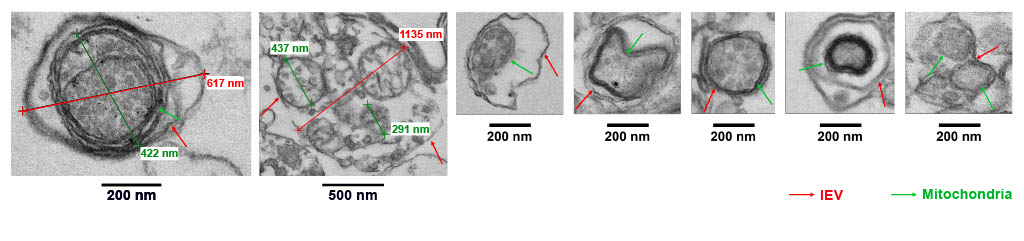
Cell-derived extracellular vesicles (EVs) carry a diverse cargo that can include mitochondria; that subset of EVs are called mitoEV. Studies have shown that EV-mediated intercellular communication is regulated, at least in part, by the EV mitochondrial cargo, suggesting the need for rigorous characterization of mitochondrial cargo load and quality. While single-particle flow cytometry holds promise in mitochondrial cargo characterization, standardized protocols and technical guidance are lacking. We developed a sensitive, reproducible, and simple protocol that employs imaging flow cytometry in combination with MitoTracker fluorescent probes to identify, quantify, and functionally assess the EV mitochondrial cargo. The protocol enables detection of mitochondrial membrane potential (ΔΨm) of mitochondrial cargo at the single mitoEV level, with ΔΨm serving as a surrogate for mitochondrial cargo quality. This platform will be useful in future studies on the role of mitoEV mitochondrial cargo quality in intercellular communication, and may lead to development of quality control standards for EV-based diagnostics and therapeutics.
Using our platform, we showed that:
- Vascular endothelial cells (ECs) release EVs with mitochondrial cargo (mitoEVs)
- ΔΨm of the EV mitochondrial cargo can be quantified using flow cytometry
- Donated EV mitochondrial cargo colocalizes with the recipient EC mitochondrial network
- EVs carrying depolarized mitochondria can induce inflammatory gene expression in recipient ECs
- A depolarized mitochondrial cargo is sufficient for mitoEVs to trigger inflammatory signaling in recipient ECs
- Antioxidants can preserve the EV mitochondrial cargo ΔΨm and protect recipient ECs from inflammation.
Hence, characterizing the circulating EC-derived EV mitochondrial cargo quality may be a new biomarker for endothelial dysfunction. Furthermore, donation of healthy mitochondria via EVs may protect vascular ECs from inflammation, and may prevent/delay CVD development. Our plans include the study of the effects of EC-derived EVs in a) hypertension, b) atherosclerosis, and c) the mitochondrial disease MELAS, using appropriate in vitro and in vivo models.
Collaborators in this project are from UB (Drs. Jennifer Lang and Jesse Slone), Roswell Park (Dr. Hans Minderman), NYMC (Dr. Sachin Gupte), Mt. Sinai (Drs. Tamas Kozicz and Graeme Preston), University of Pittsburgh (Dr. Brett Kaufman), and University of Illinois Chicago (Dr. Ele Letsiou).
Relevant publications:
- Zahid A. Manzar, Lucas J. Davis, Karl F. Swanson, Erik Muñoz, Hazel H. Szeto, Hans Minderman, and B. Rita Alevriadou, “Mitochondrial cargo quality determines the paracrine effects of extracellular vesicles derived from vascular endothelial cells”, Biomed Pharmacother, 193:118751, 2025.
Project 2:
Discovery of molecular targets for atherosclerosis through metabolomics analysis of vascular endothelial cells under atheroprone flow
Atherosclerosis leads to CVD. Systemic factors (e.g., hyperlipidemia, hypertension, baseline inflammation) are known to activate vascular ECs. EC dysfunction causes monocyte adhesion and infiltration, smooth muscle cell migration and proliferation, and eventually atherosclerotic plaque formation. However, plaques develop in arterial regions where ECs are exposed to disturbed (oscillatory) flow/shear stress (OS), not in arterial regions where ECs are exposed to undisturbed flow/shear stress (SS), suggesting that local hemodynamics plays a key role in EC dysfunction and disease development. In vivo and in vitro evidence confirmed that OS, but not SS, causes EC dysfunction.
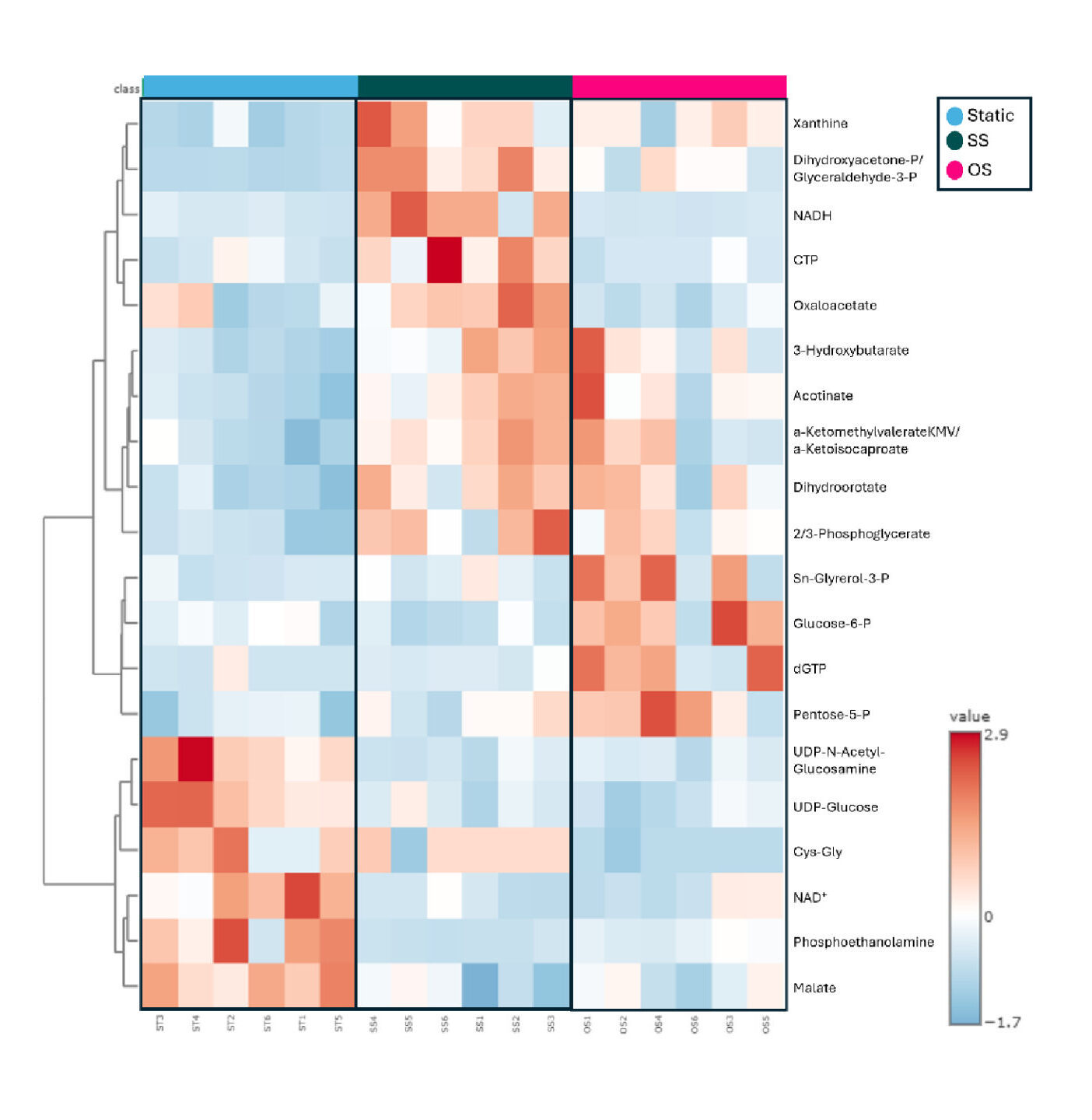
OS was also shown to increase glycolysis and decrease oxidative phosphorylation (OXPHOS), and the increased glycolysis was found to be, in part, responsible for EC dysfunction. Our goal is to better understand the shear-induced EC metabolic reprogramming, and identify molecular targets in metabolic pathways that will inhibit EC dysfunction.
Our data showed that a) SS, but not OS, protects cultured EC from inflammation in either the absence or presence of a low concentration of the proinflammatory cytokine tumor necrosis factor (TNF)-α (mimicking basal inflammation), b) ECs have an intracellular environment that is more oxidative under OS and more reductive under SS, and c) some glycolytic metabolites have significantly higher abundance under OS compared to SS. Our goal is to test pharmacological and/or genetic interventions that will prevent the OS-induced EC metabolic changes and maintain a healthy EC phenotype similar to that of SS-exposed ECs.
Understanding how shear stress regulates/reprograms EC metabolism and which changes in metabolism initiate EC dysfunction may help identify specific metabolites and metabolic pathways as targets for anti-atherosclerotic therapies.Collaborators in this project are from University of Rochester (Dr. Paul Brookes), Social Profit Network (Dr. Hazel Szeto), and UB (Drs. Stelios Andreadis and Karthik Ramachandran).
Project 3:
Sphingolipid metabolic profiles in vascular endothelial cells exposed to hypertensive vs. normotensive stretch
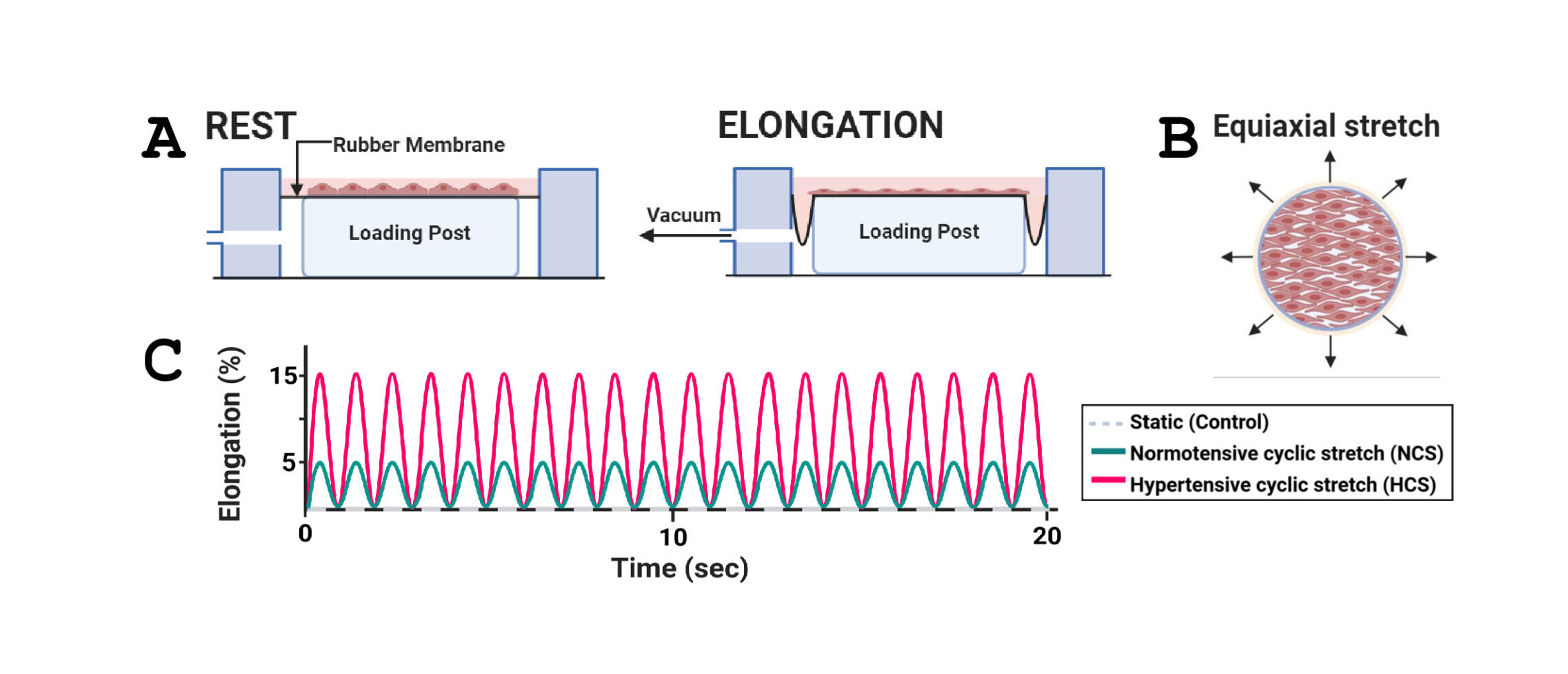
Arterial EC experience physiological cyclic stretch (CS) due to pulsatile blood pressure, which is essential for maintaining vascular function. However, excessive mechanical strain associated with hypertension can lead to EC dysfunction and the development of CVD. Cultured EC exposure to prolonged CS of high magnitude is known to cause an increase in reactive oxygen species (ROS), cytoskeletal remodeling, activation of tyrosine kinases and transcription factors, and upregulated expression of cytokines and adhesion molecules for leukocytes. Ceramides (Cer), a class of sphingolipids (SPL), play an important role in cellular processes and have been associated with hypertension. The presence of elevated Cer concentrations in plasma is considered a biomarker of CVD risk. The goal of this study is to better understand the molecular mechanisms by which alterations in SPL, in particular Cer, metabolism in cultured ECs exposed to mechanical CS regulate EC signaling, function, and survival. Cultured human ECs were either subjected to normotensive CS (NCS; 5% elongation) or hypertensive CS (HCS; 15% elongation), or left static, and SPL levels were analyzed by Liquid Chromatography-Quadrupole Time-of-Flight Mass Spectrometry (LC-QTOF-MS).
Our data showed no significant differences in Cer abundance, and increased sphingomyelin (SM) abundance, in HCS-exposed compared to NCS-exposed ECs. However, HCS caused significant increases in EC oxidative stress, inflammatory gene expression, and cell apoptosis compared to NCS. HCS-induced increases in inflammation and apoptosis were alleviated by inhibitors of the de novo Cer synthesis pathway and an inhibitor of the enzyme neutral sphingomyelinase 2 (nSMase2) that produces Cer from the breakdown of SM in the plasma membrane. Our findings suggest that HCS may cause EC dysfunction, at least in part, via Cer accumulation in specific subcellular locations, and nSMase2 may be a therapeutic target in hypertension-induced EC dysfunction and CVD. Collaborators in this project are from UB (Drs. Ekin Atilla-Gokcumen and Viviana Monje) and University of Kentucky (Dr. Erhard Bieberich).
THE AVML GROUP AND RESEARCH EFFORTS HAVE BEEN OR ARE CURRENTLY SUPPORTED BY:
- The National Institutes of Health/National Heart, Lung, and Blood Institute (NIH/NHLBI)
- R29HL054089
- R01HL067027
- R21HL091417
- R21HL106392
- R01HL142673
- The American Heart Association (AHA)
- 16GRNT27210014
- AHA Predoctoral Fellowship (Giedt)
- State Funds
- SUNY Empire Innovation Program (EIP)
- SUNY Promoting Recruitment, Opportunity, Diversity, Inclusion and Growth (PRODiG) Initiative
- Foundations
- Whitaker Foundation research grant award
- Howard Hughes Medical Institute (HHMI) MED into GRAD Predoctoral Fellowship (Scheitlin)