Research Members: Sijia Liu, Taruna Seth
Relation extraction from biomedical texts |
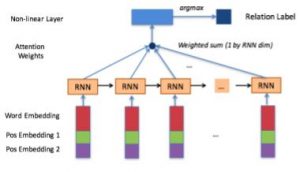
In the era of big data in biomedical domains, relation understanding among more than one entities remains one of the major challenges in biomedical Natural Language Processing (NLP) of efficient usage of unstructured texts, such as scientific literatures, clinical texts, and narrative texts in knowledge bases. The detection of relations among entities is of key relevance in machine understanding of unstructured texts. Despite the considerable number of existing systems, most of them use fine-tuned supervised machine learning methods on heavy feature engineering, which make the systems lack of generalizability and model portability. Aiming those common issues in real-world NLP applications, we proposed unsupervised learning methods extending Latent Dirichlet Allocation (LDA) to extract of coreference relations [1] and drug-drug interactions [2]. Our proposed methods can achieve comparable results to the state-of-the-art systems without using gold standard annotations. This research also addresses other challenges in biomedical natural language processing such as corpus availability, feature representation and model interpretability. We leverage various machine learning methodologies, including semantic word embedding and deep neural networks, to solve a wide range of relation extraction problems such as event temporal relations [3] and other biomedical semantic relations [4,5]. Related Publications
|
Interaction Analysis |

Characterization of pharmacological signal transductions leading to drug-induced expressions of genes and proteins requires the capability to identify interactions between different potential predictor components, e.g. genomic data, clinical data, and environmental data. Our work primarily focuses on the problem of effective characterization and detection of critical gene-gene and gene-environment interactions associated with the outcomes of interest. This problem is very challenging because the computational complexity of the interaction problem is exponential as there are n-choose-k ways of selecting a subset of k attributes for assessing interactions from n attributes. The addition of extra dimensions to a mathematical space exponentially increases the hyper volume in which data is distributed. This combinatorial growth makes it computationally difficult to exhaustively search the full range of genetic and environment (predictor) variables for potential interactions associated with diseases or outcomes in epidemiologic studies. Also, during statistical analysis of interactions using methods such as logistic regression, an additional combinatorial explosive problem arises because several different models for the data have to be evaluated to select the best model based on a suitable criterion combining goodness-of-fit and parsimony considerations.Our work primarily focuses on the problem of effective characterization and detection of critical gene-gene and gene-environment interactions associated with the outcomes of interest. This problem is very challenging because the computational complexity of the interaction problem is exponential as there are n-choose-k ways of selecting a subset of k attributes for assessing interactions from n attributes. The addition of extra dimensions to a mathematical space exponentially increases the hyper volume in which data is distributed. This combinatorial growth makes it computationally difficult to exhaustively search the full range of genetic and environment (predictor) variables for potential interactions associated with diseases or outcomes in epidemiologic studies. Also, during statistical analysis of interactions using methods such as logistic regression, an additional combinatorial explosive problem arises because several different models for the data have to be evaluated to select the best model based on a suitable criterion combining goodness-of-fit and parsimony considerations. |
Vascular Blood Flow Dynamics |
|
In this project we investigate the problem of blood flow dynamics using computational fluid dynamics (CFD) solutions. This problem is particularly cogent because of the ubiquity of 3D vascular data and as a result, a number of groups are developing patient-specific methods for calculating the flow field and flow parameters associated with vascular abnormalities via CFD. High-order CFD calculations themselves run relatively automatically, but they do require several hours on a standard computer for steady-state solutions and tens of hours for pulsatile flow solutions. Because the boundary conditions are not that well known, the reliability of a specific result for specific boundary conditions is uncertain. It might be more sensible and clinically relevant to obtain multiple CFD solutions to allow perception of trends of CFD results that correspond to the range of solutions. Unfortunately, multiple CFD solutions dramatically increase the computational complexity.We have developed CFD code which is in the process of being transferred to GPU implementation along with the conversion of our 3D CFD code. From the 2D investigations, the changes in the flow patterns with changing parameters, such as percent stenosis, length of stenosis, viscosity, and velocity, appear to follow relatively simple relationships, i.e., differential changes in the parameters result in differential changes in the flow patterns (see figure below). These results bode well for implementation of the larger strategy of interpolation based on a large data base. In addition to the CFD calculations, we are pursuing analysis of angiograms to determine whether we can determine the flow field or at least a good approximation of the flow field from high-frame-rate angiographic acquisitions as the contrast flows through the vessel. Here GPUs will be a critical component as our initial investigations involve generation and analysis of simulated angiograms for a variety of situations. Using standard CPUs, generation of single angiographic sequences take several minutes, with the analysis to calculate the flow field taking tens of minutes. We will port these calculations to GPU in the near future to allow us to evaluate the various situations investigated using CFD and to allow us to move to an iterative approach for calculation of the flow field, which will use our initial flow field calculation as a starting point. |